Hereditary Spherocytosis (HS)
Hereditary Spherocytosis (HS) was first described in 1871 and the first recorded splenectomy performed soon after. HS shows a wide range of features in regard to severity, mode of inheritance, and the actual cause of the protein defect. It is a relatively common disorder with an incidence in North Europe and North America of between 1 in 2,000 to 1 in 5,000 (depending on method of analysis). Affected patients will show a variable degree of anemia, jaundice, reticulocytosis, and splenomegaly with the clinical severity ranging from symptom free (carrier) to severe hemolysis. Exacerbation of clinical severity can be caused by illnesses such as Infectious Mononucleosis. The majority (75%) of cases will have a family history even if HS has not been formally diagnosed e.g., anemia, splenectomy, gall stones, jaundice. The diagnosis of HS has been made from birth all the way up to people in their 80’s-90’s, sometimes the diagnosis is made serendipitously. HS carriers may not show anemia, jaundice or splenomegaly and with no spherocytes seen on the blood smear.
Red Cell Morphology
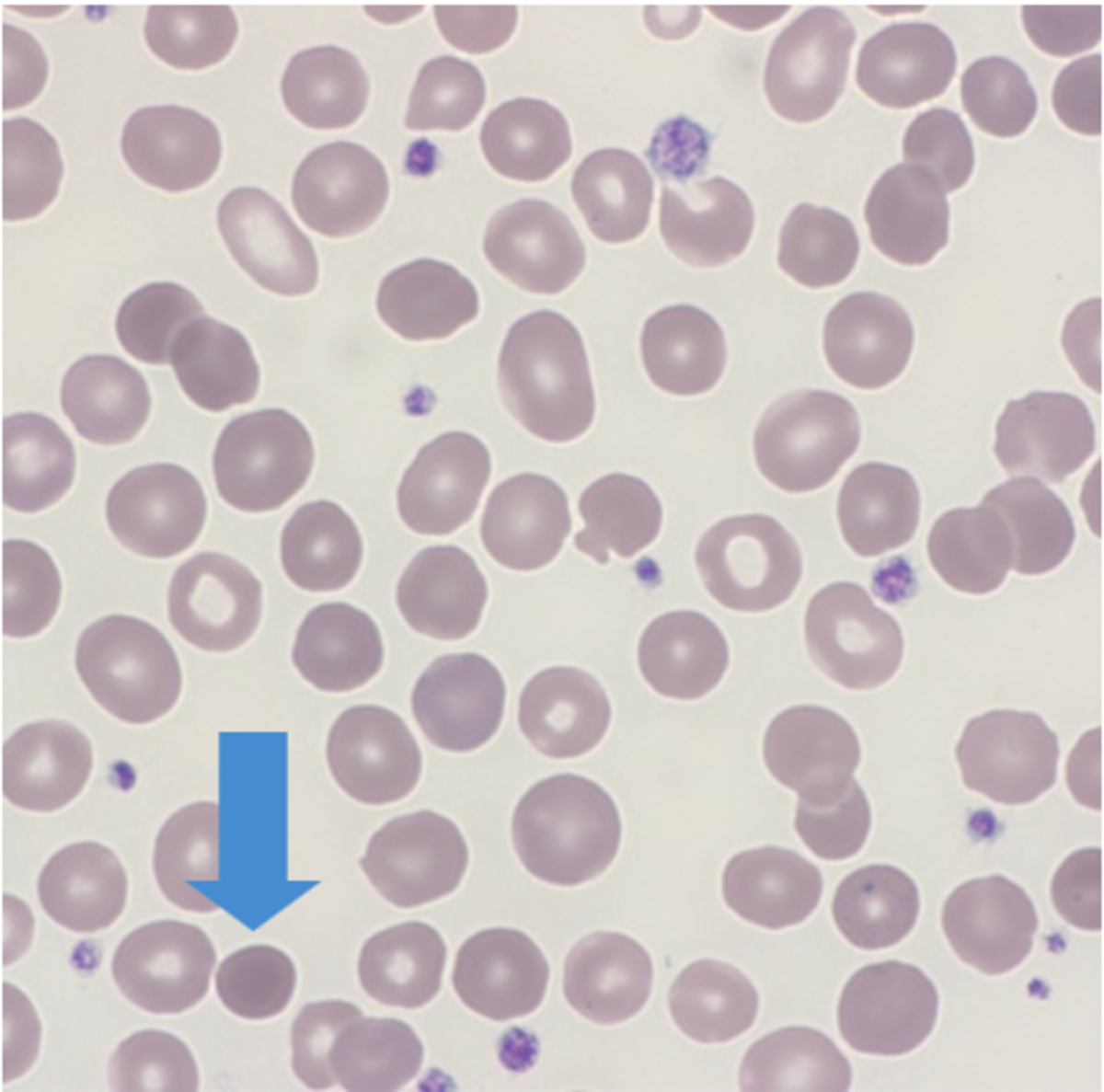
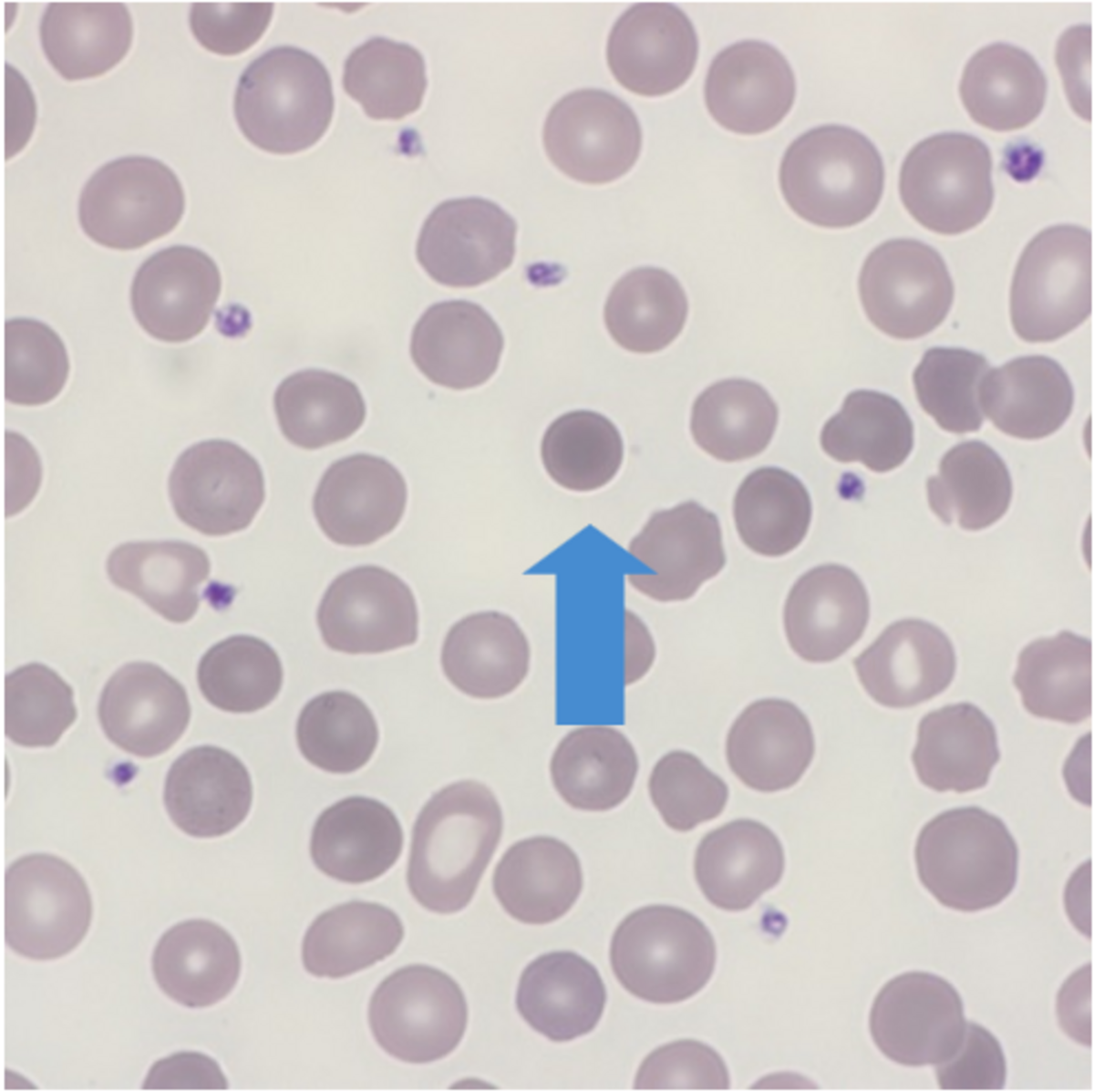
Normal red cells are biconcave discs which have a large surface area to volume and have no nucleus allowing the red cell to contain relatively large amounts of hemoglobin. Red cells need to be flexible in order to travel through tiny capillaries so that they can absorb and deliver oxygen where needed. A disturbance in the cytoskeleton can have a profound effect on the flexibility and eventual life span of the red cell. Red cell membrane integrity is dependent upon several proteins with spectrin being one of the main contributors. Abnormal red cell morphology seen in HS is due to a specific genetic mutation leading to a deficiency of, or a dysfunction in spectrin, ankyrin, band 3 and or protein 4.2.
Spherocytic red cells are removed from circulation by the spleen resulting in splenomegaly and an increase in bilirubin.